
Interest of Justice made the 4-minute presentation below at the Vaccines and Related Biological Products Advisory Committee (VRBPAC) meeting on May 22, 2025:
All credit for the information below goes to Interest of Justice.
I STRONGLY encourage the reader to support their important work: SUBSCRIBE | DONATE

https://www.pfizer.com/news/behind-the-science/how-once-little-known-molecule-disrupting-medicine
DEFINITIONS:
Human gene therapy: Human gene therapy seeks to modify or manipulate the expression of a gene or to alter the biological properties of living cells for therapeutic use.
PETITION TO THE SECRETARY OF HEALTH AND HUMAN SERVICES FOR IMMEDIATE RECLASSIFICATION OF COVID-19 mRNA AND ADENOVIRAL VECTOR PRODUCTS AS GENE THERAPY PRODUCTS
PETITION OVERVIEW:
This petition presents conclusive evidence that COVID-19 mRNA and adenoviral vector products meet the FDA’s regulatory definition of gene therapy products and must be regulated accordingly.
The Secretary of Health and Human Services possesses non-discretionary statutory duties to properly classify these products based on their functional characteristics and mechanism of action, and to address the systemic pattern of regulatory evasion that has enabled their unprecedented deployment without proper oversight.

ACTIONS REQUESTED:
Pursuant to 21 C.F.R. § 10.30, Petitioner respectfully requests that the Food and Drug Administration (FDA) immediately:
-
Reclassify all COVID-19 mRNA and adenoviral vector products as gene therapy biologics under 21 C.F.R. § 312.3(b) and 21 C.F.R. § 1271.3(d), and require all such products to comply with the regulatory requirements for gene therapy products, including but not limited to premarket review, labeling, and post-market surveillance.
-
Issue an immediate public health advisory correcting prior misclassification and informing the public and healthcare providers of the gene therapy status of these products.
-
Suspend all current and future Emergency Use Authorizations (EUAs) and Biologics License Applications (BLAs) for COVID-19 mRNA and adenoviral products until full compliance with gene therapy regulations is demonstrated.
-
Initiate enforcement actions against any manufacturer or sponsor that has failed to comply with gene therapy regulatory requirements.
-
Provide a detailed public report on the regulatory failures and corrective actions taken, including a timeline for implementation.

EXECUTIVE SUMMARY
This Petition seeks urgent regulatory action to correct the misclassification of COVID-19 mRNA and adenoviral vector products, which, by their mechanism of action and statutory definition, are gene therapy biologics.
The FDA’s failure to regulate these products as gene therapies has resulted in widespread statutory and regulatory violations, compromised informed consent, and exposed millions to unmitigated risks.
The Department of Defense’s (DoD) involvement in the development, procurement, and distribution of these products, often under emergency powers and without adherence to established safety and ethical standards, has further exacerbated these harms.
Petitioner provides detailed legal, scientific, and ethical grounds for immediate reclassification and remedial action, and pre-rebuts anticipated government defenses by demonstrating that the statutory definitions, regulatory precedents, and scientific consensus all compel the requested relief.
A. Executive Summary of Legal and Scientific Arguments
This petition presents conclusive evidence that COVID-19 mRNA and adenoviral vector products meet the FDA’s regulatory definition of gene therapy products and must be regulated accordingly. The Secretary of Health and Human Services possesses non-discretionary statutory duties to properly classify these products based on their functional characteristics and mechanism of action, and to address the systemic pattern of regulatory evasion that has enabled their unprecedented deployment without proper oversight.
This petition’s arguments are now corroborated by recent official statements from two cabinet- level officials. Defense Secretary Pete Hegseth’s April 24, 2025 memorandum explicitly characterized COVID-19 vaccines as “experimental” when announcing the reinstatement of service members discharged for refusing vaccination.
Concurrently, Health and Human Services Secretary Robert F. Kennedy Jr. is considering removing these products from the childhood vaccine schedule, stating on April 23, 2025: “The recommendation for children was always dubious… So why are we giving this to tens of millions of kids when the vaccine itself does have profound risk?”
These official Executive Branch admissions directly contradict FDA’s classification and establish the non-discretionary nature of the Secretary’s duty to act.
The scale of these products’ administration—over 5 billion doses worldwide and over 670 million doses in the United States alone—combined with the documented absence of statutorily required safeguards, creates an urgent imperative for regulatory correction. No experimental genetic technology has ever been deployed at this scale without the comprehensive safety monitoring required for gene therapy products and without fulfilling mandatory congressional reporting requirements.
Core Legal Arguments:
Definitional Violation:
COVID-19 mRNA and adenoviral vector products mechanistically function as gene therapy products under 21 C.F.R. § 600.3(h)(5) by “mediat[ing] their effects by transcription and/or translation of transferred genetic material.” The Secretary has no discretion to exempt products meeting this definition from corresponding regulatory requirements.
Pre-Pandemic Classification Admissions:
Moderna (2019 10-K) and BioNTech (2019 F-1) explicitly acknowledged in SEC filings that their mRNA technologies were classified as gene therapy by FDA prior to the pandemic. This evidentiary admission demonstrates arbitrary reclassification without required rule making. Statutory Reporting Violations: DoD and HHS have admitted through FOIA responses that they failed to generate the mandatory congressional reports required under 50 U.S.C. § 1520a before deploying biological agents to the civilian population. This statutory violation independently renders the entire program ultra vires.
Arbitrary and Capricious Action:
FDA’s exemption of functionally identical products from gene therapy regulation based solely on therapeutic intent constitutes textbook arbitrary and capricious agency action under 5 U.S.C. § 706(2)(A). As established in State Farm, an agency cannot provide an explanation that “runs counter to the evidence.”
Scientific Evidence of Gene Therapy Mechanism:
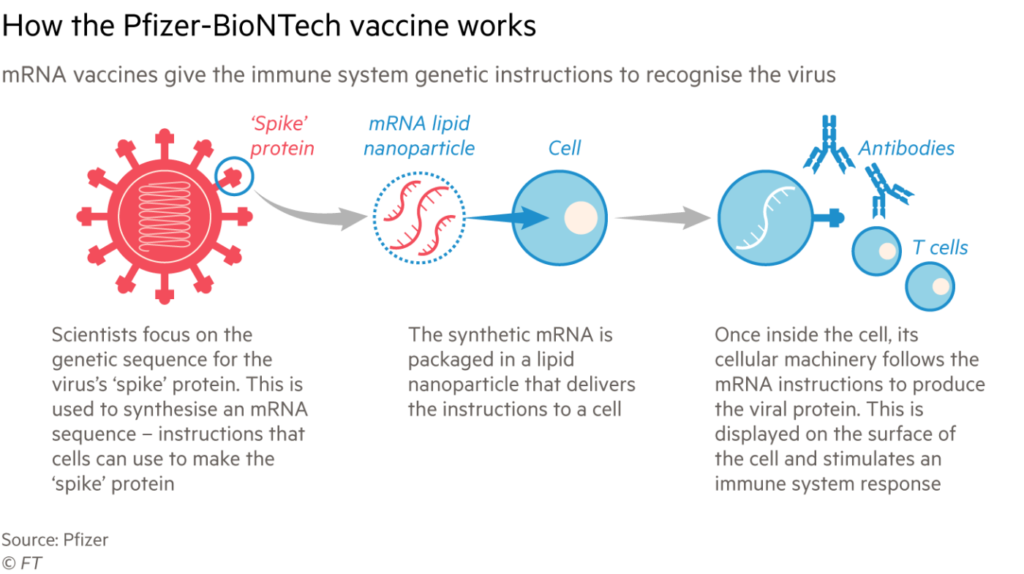
Manufacturers’ Mechanism Confirmation: Pfizer’s EMA submission describes BNT162b2 as using “mRNA that codes for the viral spike protein” which after intramuscular injection “is delivered into host cells” where “the viral spike protein is expressed.” This precise mechanism— delivering genetic material to produce non-native proteins—defines gene therapy under FDA regulations.
Extended Expression Duration:
Independent research demonstrates spike protein production for up to 60 days (Röltgen et al., Cell) and up to 15 months in lymph node germinal centers (Robertson et al., Frontiers in Immunology), confirming the “prolonged biological activity” that FDA’s gene therapy guidance identifies as requiring enhanced monitoring.
Comprehensive Biodistribution Data:
Regulatory documents confirm mRNA products distribute “rather non-specifically to several organs such as spleen, heart, kidney, lungs and brain” (EMA Assessment Report), consistent with biodistribution patterns of gene therapy products requiring enhanced safety monitoring.
Reverse Transcription Evidence:
Laboratory studies demonstrate BNT162b2 mRNA reverse transcription into DNA in human cells (Aldén et al., CIMB 2022), directly contradicting the presumption of integration impossibility used to justify exemption from genotoxicity testing.
Regulatory Violations Requiring Remedy:
Classification Violation: Failure to properly classify products meeting gene therapy definition violates FDA’s own regulations (21 C.F.R. § 600.3(h)(5)) and Secretary’s duty to ensure biological products are regulated according to their functional characteristics.
Missing Safety Testing:
Categorical waiver of genotoxicity, carcinogenicity, reproductive toxicity, and long-term expression studies required for gene therapy products violates 21 C.F.R. § 312.23(a)(8) and FDA’s gene therapy guidance.
Long-Term Monitoring Violation:
Failure to implement 5-15 year monitoring required by FDA’s own guidance for gene therapy products administered to “large number of subjects” contravenes the unreasonable risk determination standard in 21 CFR § 312.42(b)(1)(i).
Adulteration Standard Violations:
Documented DNA plasmid contamination including SV40 promoter sequences exceeding 10 ng/dose regulatory limit constitutes adulteration under 21 U.S.C. § 351 with no statutory authority for waiver.
The Secretary of Health and Human Services possesses non-discretionary statutory duty to correct these violations through immediate reclassification, clinical hold implementation, proper documentation compliance, and the other remedies detailed in this petition.
IV. LEGAL BASIS FOR RECLASSIFICATION
The Secretary of Health and Human Services possesses not merely discretionary authority but an affirmative statutory obligation under the Public Health Service Act (42 U.S.C. § 262) and the Federal Food, Drug, and Cosmetic Act (21 U.S.C. § 355) to ensure that biological products are regulated in accordance with their functional characteristics, mechanism of action, and risk profile.
The legal framework for biological product classification under the Public Health Service Act creates a non-discretionary duty for proper classification based on mechanism of action rather than therapeutic intent or administrative convenience.
The Secretary’s plenary authority under 42 U.S.C. § 262 creates an affirmative obligation to ensure that biological products are regulated according to their actual characteristics and risk profile.
The administrative law principle of substance over form, articulated by the Supreme Court in SEC v. W.J. Howey Co., 328 U.S. 293, 298 (1946), mandates that regulatory classification adhere to the functional characteristics of the regulated entity rather than nominal designation or label.
The Court has consistently reaffirmed this principle, most recently in Environmental Defense v. Duke Energy Corp., 549 U.S. 561, 574 (2007), holding that regulatory definitions must focus on “functional characteristics rather than nominal designations.”
The arbitrary exemption of mRNA products from gene therapy classification based solely on therapeutic intent rather than mechanism of action epitomizes the kind of agency action prohibited under the Administrative Procedure Act as “arbitrary, capricious, an abuse of discretion, or otherwise not in accordance with law,” 5 U.S.C. § 706(2)(A).33
A. Statutory Non-Discretionary Duty to Classify According to Functional Characteristics
The Secretary of Health and Human Services possesses not merely discretionary authority but an affirmative statutory obligation under the Public Health Service Act (42 U.S.C. § 262) and the Federal Food, Drug, and Cosmetic Act (21 U.S.C. § 355) to ensure that biological products are regulated in accordance with their functional characteristics, mechanism of action, and risk profile. This mandatory duty derives from the express statutory language, contextual framework, and underlying legislative intent of the governing statutes.
The Public Health Service Act, specifically 42 U.S.C. § 262(a)(2)(A), confers upon the Secretary the non-discretionary mandate to “establish, by regulation, requirements for the approval, suspension, and revocation of biologics licenses.” This statutory provision, interpreted according to established canons of construction, creates an affirmative obligation to ensure that biological products are classified according to their actual characteristics rather than nominal designations.
As the D.C. Circuit emphasized in Cutler v. Hayes, 818 F.2d 879, 894 (D.C. Cir. 1987), when statutory language uses the mandatory “shall,” it creates a “consequential obligation on the agency” that cannot be circumvented through administrative convenience or policy preferences.
The Supreme Court has definitively established in Massachusetts v. EPA, 549 U.S. 497, 527-28 (2007), that when statutory criteria for regulatory action are satisfied, agencies cannot decline to act based on policy considerations extraneous to the statutory framework. The Court unequivocally rejected the argument that an agency can “avoid taking further action” once a product meets statutory criteria for regulation, holding that such an approach would be “arbitrary, capricious… or otherwise not in accordance with law.” The Court specifically emphasized that when Congress has established specific criteria for regulatory classification, agencies lack discretion to create exceptions based on policy considerations.
This principle applies with equal force to the FDA’s statutory obligation to classify biological products according to their actual characteristics rather than policy preferences. When COVID- 19 mRNA products meet the regulatory definition of gene therapy products—”products that mediate their effects by transcription and/or translation of transferred genetic material”—the Secretary lacks discretion to exempt them from the corresponding regulatory framework based on policy considerations outside the statutory criteria.
The non-discretionary nature of this obligation is further reinforced by the FDA’s own binding guidance documents defining gene therapy products functionally based on mechanism of action.
Under the Accardi doctrine established in United States ex rel. Accardi v. Shaughnessy, 347 U.S. 260 (1954), agencies are legally bound to follow their own regulations and binding guidance documents.
The Supreme Court has reinforced this principle in Service v. Dulles, 354 U.S. 363, 388 (1957), holding that “regulations validly prescribed by a government administrator are binding upon him as well as the citizen.”
The Secretary’s obligation is particularly acute given the mandate in 42 U.S.C. § 262(a)(2)(B)(ii) to establish “appropriate requirements for the approval of biological products… designed to ensure the continued safety, purity, and potency of such products.” This statutory provision creates an affirmative duty to apply appropriate safety standards to biological products based on their characteristics and risks.
By exempting COVID-19 mRNA products from safety requirements that apply to functionally identical gene therapy products, the agency has violated this statutory mandate.


https://rumble.com/v6pycls-mrna-music-video-for-president-donald-trump.html

James Roguski
310-619-3055
JamesRoguski.substack.com/archive
Please help spread the word about the harmful effects of the mRNA bioweapons:


